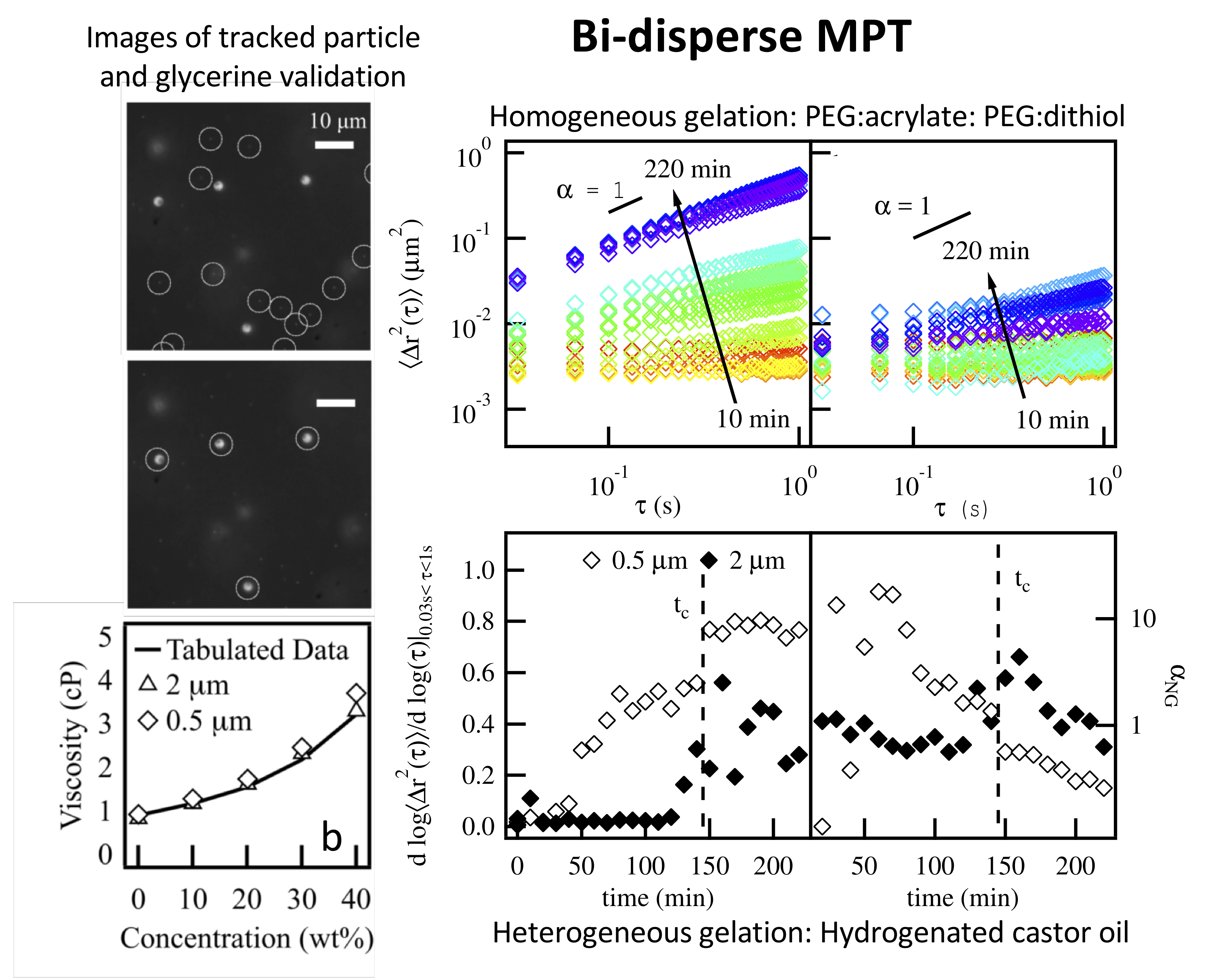
This work develops a new technique to measure several length scales in a single sample using multiple particle tracking microrheology. Traditionally, MPT uses a single particle size to characterize rheological properties. But in complex systems, MPT measurements with a single size particle can characterize distinct properties that are linked to the materials length scale dependent structure. By varying the size of probes, MPT can measure the properties associated with different length scales within a material. We develop a technique to simultaneously track a bi-disperse population of probe particles. 0.5 and 2 μm particles are embedded in the same sample and these particle populations are tracked separately using a brightness- based squared radius of gyration, Rg2.
Bi-disperse MPT is validated by measuring the viscosity of a viscous Newtonian fluid, glycerol, at varying concentrations. These measurements agree well with literature values. This technique then characterizes a homogeneous PEG-acrylate: PEG-dithiol gelation. The scaffold measured is an 18 wt% PEG-acrylate solution, which is above the overlap concentration and, therefore, includes polymeric interaction. The critical relaxation exponent, n, and critical gelation time, tc, are consistent for both particle sizes and agree with previous measurements using a single particle. Finally, degradation of a heterogeneous hydrogenated castor oil colloidal gel is characterized. The two particle sizes measure a different value of the critical relaxation exponent, indicating that they are probing different structures in the material. Analysis of material heterogeneity shows maximum heterogeneity is dependent on probe size indicating that each particle is measuring rheological evolution of a length scale dependent structure.
Overall, bi-disperse MPT increases the amount of information gained in a single measurement, enabling more complete characterization of complex systems that range from consumer care products to biological materials. Current work is using bi-disperse MPT to characterize cell-mediated pericellular remodeling and degradation in the previously discussed PEG-peptide hydrogel. During migration, human mesenchymal stem cells degrade paths in the material but must also maintain a larger scaffold structure to exert cytoskeletal tension to move. Using bi-disperse MPT, we will characterize multiple length scales within the scaffold during cell-mediated degradation. This will determine the properties of the larger structure present during hMSC motility.
Covalent adaptable
hydrogel scaffolds pushed out of equilibrium
Graduate students: Francisco Escobar IV (M. Eng. ’16) and Nan Wu
(PhD, expected ’21)
Collaborator: Prof. Kristi S. Anseth (University of Colorado at
Boulder)
Publications: N. Wu and K. M. Schultz*, “Microrheological characterization of covalent adaptable hydrogels for applications in oral delivery,” submitted
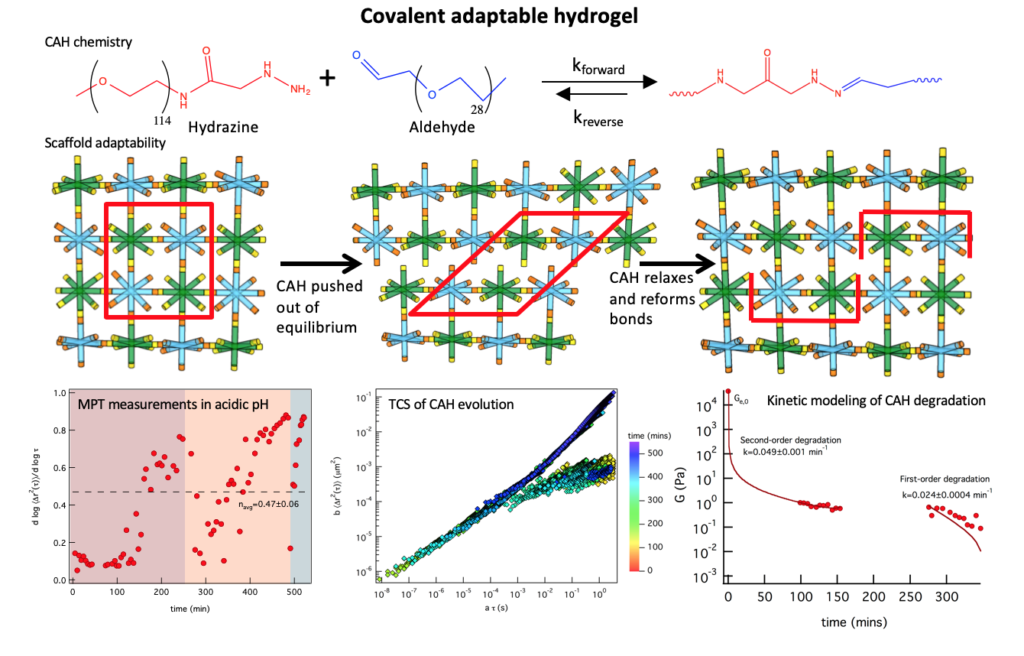
Covalent adaptable hydrogels (CAHs) mimic aspects of the native extracellular matrix cells experience in vivo due to their ability to physically adapt to their environment. The goal of this work is to measure the evolution of covalent adaptable hydrogel scaffolds, including the rheological properties and microstructure, and quantitatively link this to material function to inform the design of these scaffolds for specific biological applications. This work characterizes a covalent adaptable hydrogel developed in the Anseth group, that mimics muscle and neuronal tissue. These CAHs are made from multi-arm poly(ethylene glycol) (PEG) molecules that form reversible bis-aliphatic hydrazone bonds. This unique chemistry creates a material that yields when stress is applied and reforms covalent bonds once stress is released creating an environment that cells can survive in and responds dynamically to cytoskeletal tension during basic cellular processes. The bonds ability to break and rearrange is not only dependent on applied shear but also depends on pH, elastic moduli and equilibrium constants.
In this work, we are using multiple particle tracking microrheology to measure dynamic material properties during scaffold degradation due to a change in pH or reaction equilibrium. Microrheological measurements of this scaffold have given an abundance of information about changes that cells experience in the microenvironment during motility and cytoskeletal traction. To mimic biological processes we measure scaffold degradation in response to a change in pH (i.e. formed at a pH and incubated at a different pH). Incubation of a scaffold formed at physiological pH in an acidic buffer results in degradation of the scaffold over several hours. Interestingly, as the degradation of the material precedes we measure bonds reforming spontaneously several hours after the scaffold is pushed out of equilibrium. Using time-cure superposition (TCS) the critical degradation time, tc, and critical relaxation exponent, n, are determined during degradation. To further understand the mechanisms of this transition, we developed kinetic models of the degradation reaction. MPT was also used to characterize the scaffold when it is degraded at physiological pH. These scaffolds degrade quickly to the gel-sol transition. They then oscillate around this critical transition for about 10 days prior to complete degradation. In all, these investigations begin to build our knowledge of CAHs to inform the design of these scaffolds to enhance and encourage native biological function in the body.
Future work is continuing to characterize these scaffolds. We are
investigating the performance of these materials as an oral delivery vehicle
for medicine or nutrients in the GI tract. The GI tract has several changes in
pH, which will cause different time scales of degradation and, in turn, delivery
of encapsulated molecules throughout the process. This work a microfluidic
device that can change the incubation buffer
around the sample with minimal sample loss. Initial
work has assessed single changes in pH, from acidic to physiological. This is
similar to the change from the stomach to the intestines. These measurements
determine that the degradation time at physiological pH is accelerated when the material is first degraded at acidic pH. Future
work will mimic the temporal change in pH throughout the entire GI tract. These
measurements will then be used to tailor the scaffold to create a vehicle that
effectively delivers tethered molecules to the desired part of the GI tract and
completely degrades prior to the end of the GI tract.
Human mesenchymal stem cells (hMSCs) are critical players in wound
healing. During wound healing, hMSCs are called to the wound by chemical cues
in the environment. In response, they migrate out of their niche and traverse
mechanically distinct microenvironments to reach the wound. At the injury, they
are active in all phases of healing, regulating inflammation. hMSCs can also
restart stalled healing in chronic wounds. To enhance wound healing,
implantable synthetic hydrogels are designed to mimic in vivo microenvironments to deliver hMSCs and provide structural
integrity to the surrounding tissue. It is still not understood how cells
re-engineer their microenvironments during motility and how the
microenvironment influences cellular degradation strategies. Our approach uses
a combination of bulk rheology and passive microrheology to characterize the
bulk material integrity and pericellular region during cellular remodeling and
degradation in a synthetic hydrogel scaffold. The goal of this work is to
identify the spatial and temporal rheological evolution of hydrogel scaffolds
in response to cell-mediated degradation to determine the viability of these
materials as implantable scaffolds that enhance wound healing.
In our work, human mesenchymal stem cells are encapsulated in 3D
within a poly(ethylene glycol) (PEG)- peptide scaffold, a mimic of adipose
tissue. The scaffold used in this work has a peptide cross-linker. This peptide
is highly degradable by cell-secreted MMPs causing reproducible and predictable
degradation on the time scale of our measurements. The initial goal was to
determine if microrheology could measure cell-mediated degradation. We
successfully measured material properties of the pericellular region during
motility. In addition, the measured degradation profile was in stark contrast
to the profile generally accepted by the biomaterials community. The cell is
the source of enzymes that degrade the PEG-peptide scaffold. It was expected
that the largest scaffold degradation would occur at the cell with an increase
in cross-links further from the cell, a reaction-diffusion degradation profile.
The opposite profile was measured, a reverse reaction-diffusion profile. The
cell is protecting the material directly around it, enabling spreading and is
not motile. After spreading, the cell degrades the scaffold and has a velocity
that the average reported velocity.
From these measurements, it is clear that the cell is inhibiting
MMP degradation directly around it, which enables attachment and spreading
prior to motility. We hypothesized that hMSCs are secreting tissue inhibitors
of metalloproteinases (TIMPs) to inhibit cell-mediated scaffold degradation
directly around the cell. TIMPs bind immediately after secretion to the
catalytic portion of MMPs making them inactive. They later unbind making the
MMPs active and able to degrade the peptide cross-linker. We developed a model
using Michaelis-Menten competitive inhibition and reaction-diffusion equations
to predict whether TIMPs would be responsible for the measured degradation
profile. This model predicted that the maximum MMP-TIMP unbinding would occur
where we measure maximum degradation. Therefore, to determine the role of TIMPs
in the creation of the degradation profile, hMSCs are treated with TIMP
antibodies and MPT is used to measure 3D remodeling and degradation of the MMP
degradable PEG-peptide scaffold. After TIMP neutralization, our measurements
determine that we have achieved a reaction-diffusion degradation profile. The
change in the degradation profile also has implications on cell motility. TIMP
inhibited hMSCs have a higher motility due to either durotaxis, migration along
a stiffness gradient to stiffer regions, or reduced material barrier to motility.
This relation between cell-engineered degradation profile and hMSC motility
could be very powerful once it is well-established.
We are continuing to study the effects of both physical and
chemical cues on degradation profiles and the correlation with motility. We are
currently studying the effects of physical microenvironment changes on hMSC
degradation strategies and cellular motility. The stiffness of PEG-peptide
scaffolds is increased by increasing the cross-link density in the material. We
have measured hMSC-mediated degradation in scaffolds with stiffnesses ranging
from tens to thousands of pascals. We find hMSC degradation profile changes
from a reverse reaction-diffusion profile in soft materials to a
reaction-diffusion profile in harder materials. This is likely due to the need
for hMSCs to migrate and in order to migrate they must degrade the scaffold.
When more cross-links need to be degraded in stiffer scaffolds, the hMSCs
change their degradation strategy to a reaction-diffusion profile to enable
motility on a similar time scale as the two-step motility mechanism described
above in soft materials. This work further establishes the relation between
motility and hMSC degradation strategy.
We are also interested if
features in the physical microenvironment can be used to enhance cell delivery
to wounded areas. Present work is focusing on creating microenvironments with
rheological unique features that mimic features that hMSCs traverse when
migrating from its niche to a wound. We will create interfaces and gradients in
the material to mimic in vivo environments to measure the change in scaffold
degradation profiles in response to cell-mediated degradation. Overall, our
approach to determining cell-material inter- actions and the ability to direct
hMSC motility within the material is novel because it comes from a materials
and rheology perspective. Most studies have the goal of determining the role of
cells in these interactions. A focus on the materials and resulting rheological
properties has led to new discoveries about the environments hMSCs create
during migration and can lead to new material designs that can manipulate cellular
processes using the microenvironment.
Colloidal gels play a vital role in the stability of commercial products, such as shampoo and laundry detergent. The structure and properties of colloidal gels is dramatically changed by environmental conditions and these changes can cause decreases in shelf life and unexpected performance of the product. The goal of this work is to traverse the environmental parameter space creating a toolbox of measurement and analysis techniques that quantify dynamic colloidal gel rearrangement, degradation and heterogeneity to enhance product design, development and manufacturing.
In this work, we characterize the dynamic, heterogeneous
degradation and gelation of a hydrogenated castor oil (HCO) gel. The gel
consists of HCO fibers with surfactant crystallized along them, water, and an
environmental surfactant, linear alkylbenzene sulfonate (LAS). The surfactant
that surrounds the crystals drives bundling, and in the aqueous suspension this
results in a depletion interaction between fibers which forms a stable gel that
does not undergo gravitational collapse. The gel goes through phase transitions
in response to an osmotic pressure gradient made by contacting the scaffold
with either water or a water-starved hydrophilic liquid. In this work, we have
developed new analysis techniques that maximize information from measurements
of heterogeneous scaffold microenvironments. Material properties are determined
from the mean-squared displacements (MSDs) of probe particles temporally during
transitions. This data is analyzed using time-cure superposition to determine
the critical transition time and critical relaxation exponent, which identifies
the structure at the phase transition. Next, spatial and rheological
heterogeneities are quantified. We determine that scaffold heterogeneity does
not significantly change MPT measurements but heterogeneity analysis provides
additional insight into the evolving microstructure as the scaffold undergoes
critical transitions.
Time-cure superposition and heterogeneity analysis suggests HCO
gels return to the same equilibrium properties after repeated phase transitions.
But HCO samples that are prepared with and without shear have different
critical relaxation exponents, indicating the structure of the materials are
different. We developed a new microfluidic device that enables fluid exchange
around the sample (even when it is a sol) by using pressure to ‘lock’ it in
place. During solvent exchange around the sample, minimal shear is added to the
material. After fluid exchange, MPT is used to characterize the scaffold. With
this device we can induce several phase transitions on a single sample to
determine whether colloidal rearrangement or shear added during sample
preparation is responsible for the change in scaffold structure measured during
single gelation and degradation experiments. These measurements confirm HCO
returns to the same equilibrium properties after each phase transition,
suggesting the addition of shear during sample preparation is changing the
scaffold structure. Bulk rheological investigations of both gelation and
degradation confirm that shear added during sample preparation changes the
accessible material properties. From these studies, we find that processing
prior to material use changes the rheological properties of this rheological
modifier, which should be considered during manufacturing.
Cross-linked gels have played a significant role in enhanced oil recovery. These materials are used to decrease permeability in high permeability zones near naturally fractured carbonates that require water shutoff but cannot be permanently plugged. The goal of this work is to establish a fundamental understanding of how the interplay between polymeric interactions and cross-linking changes assembly, final structure and properties and overall stability of the gel network. These history-dependent systems are monitored during gelation to establish a quantitative framework to understand how polymeric interactions, i.e. overlap and entanglement, within macromer solutions change the gelation reaction and influence final material properties in chain- and step-growth systems. A combination of bulk rheology and microrheology is used to determine the critical values of these gels during phase transitions and the final material properties.
In this work, both
polymeric interactions and reaction mechanisms are varied to determine the
change in the gelation reaction and final material properties. Polymeric
solutions have three regimes in the concentration- viscosity curve, the dilute,
semi-dilute and entangled. In the dilute regime, polymers do not interact. In
the semi-dilute regime, the pervaded volume of the polymers begin to overlap.
In the entangled regime, polymers are entangled. In addition to changing
polymeric interactions both chain- and step-growth gelation are characterized.
The step-growth gel characterized is a four-arm star PEG-acrylate (Mn 20
000 g/mol) cross-linked with a linear PEG-dithiol (Mn 1 500). This
is a photopolymerization reaction that uses lithium phenyl- 2,4,6-trimethylbenzoylphosphinate
as a photoinitiator. The chain-growth reaction is a four-arm star PEG-
maleimide (Mn 20 000 g/mol) that self assembles with a linear
PEG-dithiol (Mn 1 500 g/mol). The
backbone and cross-linker for each gel is the same size, therefore, at the same
concentration any variation in structure is due to the mechanism of gelation.
These scaffolds are characterized using a combination of bulk rheology and
microrheology.
The logarithmic slope of
the mean-squared displacement, α, shows the change in material properties
during scaffold gelation. The step-growth gelation proceeds with a consistent
decrease in α throughout the gelation reaction. This indicates that cross-links
are constantly forming after the initiation of gelation. For a chain-growth gel
there is an initiation step which slows the onset of gelation. After enough
radicals have built up in the system gelation occurs, indicated by the sharp
decrease in α.
Time-cure superposition (TCS) is used to analyze both of these gel scaffolds at
concentrations above and below the overlap concentration. The critical
relaxation exponent indicates the structure of the network and the ability for
the network to store and dissipate energy. We determine the critical relaxation
exponent, n, decreases with
increasing concentration, indicating a change from a percolated (c < c*) to a tightly
cross-linked network (c* < c). The gelation
mechanism does not have a measurable effect on the scaffold structure.
Current work on this
project is collecting data on several other polymer concentrations around c*. This data shows that
the value of n remains constant
below and above c*. Therefore, below c* all gels, regardless of
concentration have a percolated network and above c* have a tightly
cross-linked network. The change between these network structures is a step
change.